

Dr. Stéfano Gonçalves Jorge
INTRODUÇÃO
Descrita inicialmente por Addison e Gull em 1851 e Hanot em 1876, inicialmente como uma cirrose relacionada a maior icterícia e depósito de gordura na pele (xantomas), a Colangite Biliar Primária (CBP) é uma doença presumivelmente autoimune que afeta principalmente mulheres entre 40-60 anos e, se não tratada, leva a cansaço crônico, coceira generalizada, icterícia e, em fases mais avançadas, até a cirrose e suas complicações.
Antes do surgimento do tratamento com ácido ursodesoxicólico (UDCA), a evolução para cirrose acontecia com a maioria dos portadores. Depois do UDCA, os que respondem ao tratamento tem a mesma expectativa de vida que o restante das pessoas. Quanto mais cedo for feito o diagnóstico, maior a chance de sucesso do tratamento, de levar uma vida normal e sem a necessidade de transplante de fígado.
A QUESTÃO DO NOME
Entre 1850 e 1950, a condição ficou mal definida até que foi abordada de modo mais sistemático por MacMahon em 1949 e novamente por Arens em 1950, que finalmente definiu o nome da doença como cirrose biliar primária. Esse nome persistiu por décadas, apesar de protestos da comunidade médica, já que nem sempre a pessoa tinha com desenvolvia cirrose. Mas foi só na década de 1980, com o início do tratamento com o ácido ursodesoxicólico (UDCA), que a história natural da doença realmente começou a mudar. De um lado os médicos se queixavam de que a maioria dos portadores não teriam mais cirrose, de outro os grupos de portadores queriam fugir do estigma do diagnóstico de cirrose, que só era aplicável a uma minoria.

Só que mudar nome de doença não é tão fácil, e ficou pior nas últimas décadas. Grupos de portadores, inicialmente criados para apoio comum, e depois para advogar em prol de medicamentos em benefícios, usaram a sigla CBP (ou PBC, em inglês) e não queriam mudar isso. Só entre 2014 e 1015 chegamos a um acordo de mudar o nome para colangite biliar primária, mantendo a sigla mas sacrificando um pouco a lógica.

Claro, toda colangite (inflamação nos canais da bile) é, obviamente, biliar e ficamos com um nome com pleonasmo. Por isso que ainda encontramos muita informação com o nome de cirrose biliar primária, e esse continua o nome na tabela de CID (Cadastro Internacional de Doenças).
EPIDEMIOLOGIA
É uma doença incomum, mas não rara, que acomete todas as etnias e principalmente mulheres (são 10 mulheres para cada homem afetado) entre as quarta e sexta décadas de vida. A CBP tem incidência (novos casos) de cerca de 10 a 12 casos por ano por 1 milhão de pessoas e prevalência (número total de doentes) de 100 a 150 casos por milhão. Como a maioria das doenças crônicas do fígado, a CBP pode ser assintomática e seu diagnóstico só é realizado tardiamente, já no estágio de cirrose, a não ser que seja suspeitada ou detectada precocemente por exames de rotina (especialmente fosfatase alcalina e gama glutamiltransferase). Apesar de incomum, a CBP é responsável por cerca de 0,6 a 2,0% das mortes por cirrose no mundo.
FISIOPATOGENIA
Já foram feitas muitas tentativas de se encontrar a causa dessa doença. A hipótese de que exista pelo menos uma base genética é apoiada pelo risco aumentado entre irmãs, gêmeas e mãe e filha. Estudo recente mostrou que em gêmeos monozigóticos (idênticos geneticamente), há uma taxa de concordância de 0,63, que é a mais alta já observada entre as doenças consideradas autoimunes. No entanto, ainda não foi isolado gene ou conjunto de genes claramente responsáveis pela CBP.

Há estudos sugerindo também a presença de um fator ambiental, que não foi identificado. Esse fator poderia ser uma causa direta (agentes tóxicos, como tabagismo) ou como “gatilhos” da doença autoimune. Essa hipótese está apoiada na distribuição “em grupos” da doença (apoiando a possibilidade de outros fatores, como a composição da microbiota intestinal) e em relatos de CBP relacionados a infecções, que podem tê-las desencadeado. A razão da preferência por mulheres também não foi explicada, pois quando homens manifestam a CBP ela tem exatamente as mesmas características.

É evidente, no entanto, que a doença surge com (ou por) anormalidades no sistema imunológico, que passa a destruir os canalículos biliares, sugerindo que trata-se de uma doença autoimune. Ela é frequentemente encontrada em associação com outras doenças autoimunes, como tireoidites. Além disso, a presença de anticorpo anti-mitocôndria é quase constante nos portadores (o que não significa que a doença seja causada por esse anticorpo). Vários agentes infecciosos, vírus e bactérias, já foram “acusados” de ser gatilhos desencadeadores do erro no sistema imunológico, mas nenhum de forma conclusiva.
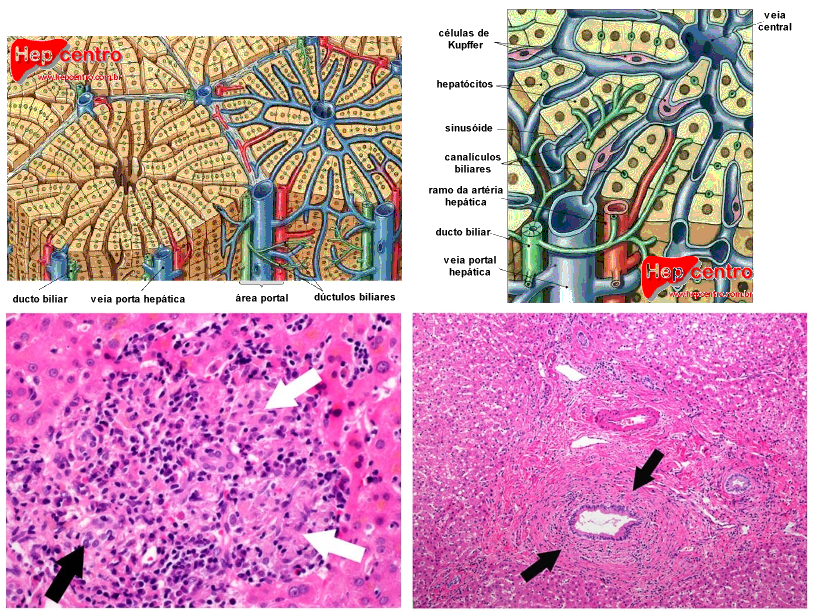
Na CBP, ocorre a destruição dos canais biliares possivelmente pelas células de defesa do tipo linfócitos T (Th1 e Th2), por mecanismo não esclarecido (mas envolvendo apoptose) e levando a redução do fluxo de bile e ao seu acúmulo (colestase). A bile acumulada causa ainda mais dano aos canais biliares e aos hepatócitos, que são lentamente destruídos com a formação de fibrose e, a longo prazo, a cirrose se estabelece. Sugere-se ainda que uma mutação genética aumentaria a expressão de endotelinas em resposta a agentes externos (como vírus), que levariam a ativação das células de Kupffer e esteladas, que por sua vez contrairiam vasos peribiliares, levando a dano das vias biliares, apoptose da mesma e exposição de antígenos mitocondriais que levam à produção do anticorpo antimitocôndria (que, portanto, não seria autoanticorpo responsável pela doença, mas um criado a partir dela e que levaria a dano adicional).
SINTOMAS
O sintoma mais frequente (cerca de 80%) da CBP é fadiga, cuja intensidade não tem relação com a atividade ou o estágio da doença. Não sabemos bem por que ela ocorre, mas há indícios de que a colestase (acúmulo de bile) afete o sistema nervoso central e cause disfunção da musculatura periférica, levando a disfunção autonômica, sonolência diurna, distúrbio no sono noturno, dificuldade de concentração, problemas de memória e depressão.

O segundo sintoma em frequência (20-70%) é o prurido (coceira), o que pode levar a pessoa a procurar inicialmente um dermatologista. A icterícia (amarelão) pode estar ausente, pois não é a bilirrubina que causa a coceira, mas outras substâncias (sais biliares, autotaxina, ácido lisofosfatídico) que são normalmente excretadas na bile mas graças à colestaseacabam sendo absorvidas para a circulação.
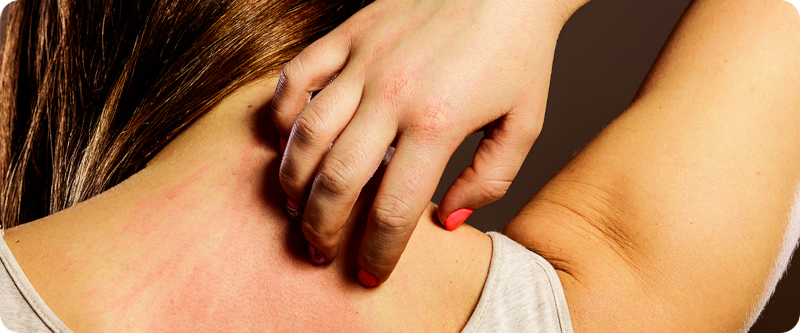
Outro sintoma característico, que independe da gravidade da CBP e pode surgir ou desaparecer é o xantoma, depósitos de gordura sob a pele, devido ao aumento do colesterol total no sangue (que não trata-se do colesterol “normal”, mas de uma lipoproteína anômala produzida em pacientes com colestase e que não está associada a aumento do risco cardiovascular).

Portadores frequentemente apresentam também outras doenças autoimunes, particularmente a síndrome de Sjogren (doença autoimune que ataca as glândulas salivares e lacrimais, levando a boca e olhos cronicamente secos). Felizmente, a presença de outras doenças autoimunes não piora a evolução da CBP.
| CBP e doenças autoimunes associadas | |
| Ceratoconjuntivite seca | 50% |
| Síndrome de Sjogren | 3.5–73% |
| Artrite reumatóide | 5,6-10% |
| Tireoidite de Hashimoto | 5,6% |
| Fenômeno de Raynaud | 4,0-9,0% |
| Escleroderma | 1,4-2,0% |
| Colite ulcerativa | 0,3% |
DIAGNÓSTICO
O diagnóstico da CBP é geralmente feito em quatro situações:
- após exames de rotina, onde observou-se elevação de fosfatase alcalina e/ou colesterol total, sendo então a pessoa submetida a investigação da causa dessas alterações;
- pessoas portadoras de outras doenças imunológicas que estão associadas a um risco maior de CBP (doenças da tireóide, escleroderma, artrite reumatóide) e são investigadas;
- pacientes com osteoporose secundária à colestase crônica, principalmente se associada a menopausa sem reposição hormonal;
- pacientes já com cirrose, sendo que nessa fase o diagnóstico da CBP nem sempre é possível.
| Diagnóstico da Cirrose Biliar Primária | ||
| Clínica | Mulher de meia-idade | 90% dos pacientes |
| Fadiga | 60-70% dos pacientes | |
| Prurido (coceira) | 50-60% dos pacientes | |
| Hepatoesplenomegalia | ||
| Outras doenças autoimunes | ||
| Xantomas | ||
| Sinais de cirrose | ||
| Sem sintomas | 30-40% dos pacientes | |
| Bioquímica | Elevação de fosfatase alcalina | 3 ou mais vezes acima do normal |
| Elevação de gama-glutamil-transpeptidase | ||
| Elevação do colesterol | 80% dos pacientes | |
| Bilirrubina direta normal ou elevada | normalmente pouco aumentada | |
| Sorologia | Anticorpo-antimitocôndria (título > 1:40) | 90-95% dos pacientes* |
| IgM elevado | 95% dos pacientes | |
| Radiologia | Colangiopancreatografia retrógrada endoscópica normal | normalmente é desnecessária |
| Histologia | Destruição granulomatosa de ductos biliares | |
| Proliferação de ductulos biliares | ||
| Infiltração crônica por células inflamatórias | ||
| Fibrose com ou sem cirrose | ||
A fosfatase alcalina está aumentada em quase todos os casos. Como é produzida não só nas vias biliares, o seu aumento isolado (sem aumento concomitante da gama glutamil transferase) pode ser devido a lesões em outros órgãos (como os ossos). A eletroforese da fosfatase alcalina permite detectar qual das isoenzimas está aumentada e direcionar a investigação se não houver sinais de doença hepática. Tipicamente, as transaminases (AST e ALT) podem estar normais ou pouco elevadas.

O anticorpo anti mitocôndria (AMA) está presente em cerca de 90-95% dos portadores, especialmente o AMA subtipo M2. Dos poucos portadores com AMA negativo, a grande maioria tem sorologia FAN positiva, especialmente nos padrões gp210 e sp100, que indicam doença mais grave.
A única lesão diagnóstica da cirrose biliar primária no estudo histológico da biópsia hepática é a destruição dos ductos biliares septais ou interlobulares com a formação de granulomas, mas esse achado dificilmente é encontrado em biópsias por agulha, pelo tamanho limitado da amostra. As alterações normalmente encontradas são divididas em 4 estágios:
| Estágios Histológicos da Cirrose Biliar Primária | |
| I | ou portal, é caracterizada por inflamação dos tratos portais ao redor dos ductos biliares |
| II | caracterizado por maior destruição biliar, proliferação dos dúctulos biliares (ductos menores), necrose em “saca-bocado” e progressão da inflamação das áreas portais para o parênquima hepático |
| III | com fibrose se estendendo dos tratos portais |
| IV | ou septal, definido pela presença de nódulos regenerativos delimitados por fibrose |

Apenas o estágio I seria mais característico da CBP, pois os demais podem ser observados em outras doenças hepáticas crônicas. Com exames laboratoriais tão específicos, é até discutível o papel da biópsia no diagnóstico da CBP. Mesmo em relação ao estágio da doença, o valor da biópsia hepática é limitado pois a doença apresenta estágios histológicos diferentes no mesmo fígado.
| Diagnóstico Diferencial |
| Obstrução biliar (cálculos, estenoses ou tumores) |
| Colangite esclerosante primária |
| Colestase induzida por drogas |
| Sobreposição com hepatite autoimune (overlap syndrome) |
| Sarcoidose |
| Ductopenia idiopática do adulto |
| Colangite autoimune |
Exames de imagem tipicamente são pouco úteis para o diagnóstico da CBP, mas são necessários em todos os pacientes. Ultrassom, tomografia (com contraste) ou ressonância nuclear magnética (com contraste) servem para descartar nódulos, mostrar sinais de doença crônica ou cirrose e se há dilatação das vias biliares, que pode sugerir a presença de outras doenças, como cálculos ou tumores. Nem sempre esses exames mostram bem o estágio da doença, podendo-se associar exames de elastografia transitória ou ultrassônica. A colangioressonância é o exame de escolha para investigar as vias biliares, e geralmente é normal na CBP pois essa afeta apenas os canalículos biliares (os mais finos). Por outro lado, ajuda a diferenciar a CBP da colangite esclerosante primária, que afeta os canais maiores e aparece na colangioRM.
O diagnóstico da CBP, na prática, é feito com 2 dos 3 critérios abaixo:
- aumento de enzimas canaliculares (fosfatase alcalina e gama glutamil transferase)
- anticorpo antimitocôndria positivo (ou, alternativamente, aumento de IgM e presença de FAN nos padrões gp210 e sp100)
- biópsia hepática mostrando lesão de canalículos biliares
HISTÓRIA NATURAL
Até hoje a história natural da CBP não está bem estabelecida, mas está clara a tendência a evolução com cirrose e suas complicações, (incluindo ascite, varizes esofágicas e encefalopatia hepática). Um estudo mostrou que a sobrevida média de pacientes assintomáticos era de 16 anos após o diagnóstico e de 8 anos para os sintomáticos, mas isso antes de existir o tratamento com ácido ursodeoxicólico. Como toda cirrose, a causada pela CBP aumenta o risco de desenvolver hepatocarcinoma, mas esse risco é mais evidente em homens.
| Complicações da CBP | |
| Pela cirrose | hipertensão portal |
| sangramento de varizes esôfago-gástricas | |
| ascite | |
| encefalopatia hepática | |
| Pela colestase | osteopenia / osteoporose |
| deficiência de vitaminas lipossolúveis (A, D, E e K) | |
| hipercolesterolemia | |
Não há nenhum método confiável para estimar o tempo de vida de um portador de CBP (o mais estudado, com resultados questionáveis, é o Mayo Risk Score). Observou-se que portadores de uma cirrose micronodular (com nódulos pequenos) evoluem mais rapidamente do que os com cirrose macronodular. Dentre todas as características possíveis para essa estimativa, o valor de bilirrubina total é o mais importante. A bilirrubina pode permanecer normal por vários anos e depois permanecer pouco aumentada por mais alguns. Quando começa a surgir falência do fígado, o nível de bilirrubina sobe muito, especialmente nos últimos dois anos de vida, tornando essa mudança (bilirrubina total acima de 4-6 mg/dL) um bom método para encaminhar o paciente ao centro de transplante hepático.
TRATAMENTO
Estilo de vida
O tabagismo está associado a progressão da doença, portanto o recomendável é parar de fumar. O consumo de álcool tende a potencializar lesão no fígado independente da dose e da doença, portanto a abstinência alcoólica também é recomendável. Não há uma relação direta entre obewsidade e piora da CBP, mas o ideal é manter o -peso dentro do adequado, além de dieta balanceada e atividade física adequadas à idade.
Sintomas
A fadiga é um fenômeno mal compreendido, já que não tem relação direta com a atividade ou o estágio da doença. Assim, não temos um tratamento adequado no momento. O único tratamento promissor atualmente é o modafinil, um estimulante desenvolvido para tratamento de narcolepsia. A recomendação é sempre pesquisar e tratar outras causas de fadiga, como anemia, hipotireoidismo, depressão e distúrbios do sono.

A causa do prurido ainda é incerta, pois não está diretamente relacionada ao nível sanguíneo de bilirrubina. Mesmo assim, o uso de um quelante de sais biliares (substância que se liga aos sais biliares e os impede de ser reabsorvidos pelo sangue), a colestiramina (questran®), em uma dose de até 4 a 16 gramas por dia, é a primeira escolha no tratamento. Pode causar gases e constipação, mas a principal preocupação é alterar a absorção de outras drogas, especialmente o UDCA. Por esse motivo, outros medicamentos tem que ser tomados pelo menos 1 hora antes ou 4 horas após a colestiramina.

Quando não há resposta, é possível o uso de rifampicina, iniciando-se com 150 mg duas vezes por dia e dobrando a dose se não houver melhora. Infelizmente, pode ser tóxica ao fígado, ao rim e causar anemia hemolítica, portanto esses efeitos colaterais tem que ser bem monitorados. Outros tratamentos, menos utilizados pelo perfil de complicações ou baixa eficácia incluem a naltrexona e a sertralina.
A colestase crônica tende a reduzir a absorção de enzimas lipossolúveis no intestino, que depende do fluxo adequado de bile. Assim, há risco aumentado de osteoporose ou outras complicações a longo prazo, que são preveníveis com a reposição adequada.
| Sintomas relacionados à deficiência de vitaminas lipossolúveis* | |
| Vitamina A | cegueira noturna (excesso pode ser tóxico ao fígado) |
| Vitamina D | osteoporose, osteomalácia |
| Vitamina E | alterações neurológicas |
| Vitamina K | coagulopatia (sangramentos espontâneos) |
Xantomas múltiplos podem levar a dor por infiltração dos nervos, podendo ser tratada com plasmaférese (“filtração” do sangue). Os olhos secos melhoram com colírios que simulam a lágrima. Fenômenos de Raynaud podem ser tratados com bloqueadores de canais de cálcio.
À medida que a doença progride, podem surgir as complicações esperadas para todas as doenças hepáticas crônicas. No caso da CBP, como ocorre fibrose pré sinusoidal, a hipertensão portal pode se desenvolver antes da cirrose, com todas as suas complicações (ascite, hemorragia por varizes, encefalopatia hepática, etc.). Do mesmo modo, após a cirrose há o risco de desenvolver hepatocarcinoma, que por algum motivo desconhecido é duas vezes mais provável em homens do que em mulheres com CBP.
Prevenção
Osteoporose e osteopenia podem ser em parte prevenidos pela suplementação de vitamina D e cálcio, além de banhos de sol mínimos. O uso de hormônios em mulheres menopausadas pode ajudar, mas a dose deve ser mínima (e preferencialmente em adesivos), pois o estrógeno pode piorar a colestase e a lesão hepática.
O risco de sangramento por varizes de esôfago pode ser reduzido com o rastreamento prévio de varizes e a profilaxia com drogas (propranolol, carvedilol) ou ligadura elástica de varizes, ou ambos. Atualmente, há preferência para a profilaxia primária (antes do primeiro episódio de sangramento) apenas com medicamento, e a secundária com a associação com a ligadura elástica.
Como a hipercolesterolemia é causada por lipoproteína anômala (lipoproteína X) que não está associada a risco cardiovascular, não está claramente indicado o tratamento com drogas hipolipemiantes, embora o uso desses medicamentos possa em teoria reduzir a progressão para a cirrose.
Tratamento medicamentoso
O ácido ursodeoxicólico – UDCA (ursacol ®) é ácido biliar que tem a característica de ser mais hidrofílico (mais solúvel em água) que a maioria dos sais biliares normalmente presentes na bile. Quando administrado em doses terapêuticas (13 a 15 mg/kg/dia), passa a constituir cerca de 40% do pool de ácidos biliares na bile, que torna-se menos detergente e tóxica. Além disso, parece ter efeito mudando aspectos de imunidade nos hepatócitos e canais biliares, estimulando a secreção hepatobiliar (reduzindo a colestase) e tem uma ação anti-apoptótica de mecanismo ainda não adequadamente esclarecido. Há melhora significativa, tanto laboratorial quanto à biópsia hepática, na maioria dos pacientes, que provavelmente terão progressão mais lenta da doença.

Os estudos realizados sobre o benefício do UDCA a longo prazo na CBP, evidenciam redução na progressão da doença, óbito ou necessidade de transplante. Acredita-se que o tratamento seja especialmente eficaz na fase pré-cirrótica, quando os benefícios em termos de progressão da doença são mais acentuados. Um estudo recente mostra que, se em 6 meses de tratamento, o portador mantiver a fosfatase alcalina acima de 1,9 vezes o limite do laboratório indica que muito provavelmente não responderá adequadamente ao tratamento ao final de 1 ano e portanto já podemos passar para uma segunda opção de tratamento.

O ácido obeticólico (OCA) é um ácido biliar hidrofílico semi sintético que atua diretamente nos genes envolvidos no metabolismo de ácidos biliares e que é considerado tratamento de segunda linha nos não respondedores ao UDCA. A dose inicial é de 5 mg por dia, que será aumentada para 10 mg/d após 6 meses. Estudos mostram excelentes resultados em termos de normalização das enzimas canaliculares de pacientes que não responderam ao UDCA, mas essa nova medicação foi relacionada a piora na fadiga e no prurido, levand a suspensão do tratamento em até um terço dos pacientes dependendo da dose. Tambpem observou-se aumento de problemas cardíacos por algum mecanismo não esclarecido. Essa medicação ainda não está disponível no Brasil, espero que até que seja liberada saibamos como evitar os efeitos colaterais.
Os fibratos (bezafibrato e fenofibrato) também tendem a aumentar os níveis das enzimas canaliculares em portadores de CBP que não respondem bem ao UDCA sozinho. No Brasil associar o UDCA com um fibrato ainda é a segunda linha de tratamento, independente de qual (não está claro qual é mais eficaz ou mais seguro). Apesar de melhorar os exames, não está bem claro se melhoram a evolução da doença a longo prazo, e se o risco de toxicidade renal ou hepática valem a pena, especialmente em quam já tem cirrose.
A budesonida, um corticosteóide com poucos efeitos colaterais sistêmicos, é outra medicação que pode ser associada ao UDCA em não respondedores. Pode trazer melhora nos exames laboratoriais, mas aparentemente não na biópsia hepática, o que significa que pode não alterar a evolução da doença. O que foi observado em estudos até agora é que os efeitos colaterais, incluindo trombose portal temdem a ocorrer em pacientes cirróticos, portanto no momento está contra indicada em portadores de cirose.
Transplante hepático
O transplante hepático é a melhor opção para portadores de CBP em estágio avançado. A probabilidade de sobreviver ao 1o. ano é de 90 a 95% em grandes centros e de 70 a 80% em 5 anos. Há uma taxa de recidiva de cerca de 40% da CBP no fígado transplantado em 15 anos, mas nesses a doença costuma seguir um curso mais benigno. As indicações mais aceitas para transplante são:
- bilirrubina total acima de 4 mg/dL
- falência da função hepática (MELD-Na acima de 15 pontos)
- prurido ou fadiga graves e refratárias
- ascite refratária ao tratamento
- encefalopatia hepática
- sangramento por varizes não controlado por medicação e endoscopia
BIBLIOGRAFIA
- Addison T. Gull W. On a certain affection of the skin – vitiligoidea a plana, b tuberosa. Guy’s Hospital Reports. 1851; 7: 265-276
- Hanot V. Étude sur une forme de cirrhose hypertrophique du foie [cirrhose hypertrophique avec ictere chronique]. JBBaillère, Paris, 1876
- MacMahon H.E. e Thannhauser S.J. Xanthomatous biliary cirrhosis: a clinical syndrome. Ann Intern Med. 1949; 30: 121-179
- Ahrens Jr., E.H. Payne M.A. Kunkel H.G. Eisenmenger W.J. Blondheim S.H. Primary biliary cirrhosis. Medicine. 1950; 29: 299-364
- Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, Ma X, Mackay IR, Parés A, Tanaka A, Vierling JM, Poupon R. Changing Nomenclature for PBC: From ‘Cirrhosis’ to ‘Cholangitis’. Am J Gastroenterol. 2015 Nov;110(11):1536-8. doi: 10.1038/ajg.2015.312. Epub 2015 Sep 29. PMID: 26416194; PMCID: PMC4679751.
- You H, Duan W, Li S, Lv T, Chen S, Lu L, Ma X, Han Y, Nan Y, Xu X, Duan Z, Wei L, Jia J, Zhuang H; Chinese Society of Hepatology, Chinese Medical Association. Guidelines on the Diagnosis and Management of Primary Biliary Cholangitis (2021). J Clin Transl Hepatol. 2023 Jun 28;11(3):736-746. doi: 10.14218/JCTH.2022.00347. Epub 2023 Feb 10. PMID: 36969891; PMCID: PMC10037524.
- Sherlock, S e Heathcote, J em Bircher, J, Benhamou, JP et al. Oxford Textbook of Clinical Hepatology, Oxford Medical Publications,1999.
- Lindor, Keith D.*; Bowlus, Christopher L.; Boyer, James; Levy, Cynthia; Mayo, Marlyn. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 69(1):p 394-419, January 2019. | DOI: 10.1002/hep.30145
- Lewis J. Pathological patterns of biliary disease. Clin Liver Dis (Hoboken). 2017 Nov 30;10(5):107-110. doi: 10.1002/cld.667. PMID: 30992767; PMCID: PMC6467117.
- Song P, Zhang X, Feng W, Xu W, Wu C, Xie S, Yu S, Fu R. Biological synthesis of ursodeoxycholic acid. Front Microbiol. 2023 Feb 24;14:1140662. doi: 10.3389/fmicb.2023.1140662. PMID: 36910199; PMCID: PMC9998936.
- Zhang Q, Liu Z, Wu S, Duan W, Chen S, Ou X, You H, Kong Y, Jia J. Meta-Analysis of Antinuclear Antibodies in the Diagnosis of Antimitochondrial Antibody-Negative Primary Biliary Cholangitis. Gastroenterol Res Pract. 2019 Jun 10;2019:8959103. doi: 10.1155/2019/8959103. PMID: 31281353; PMCID: PMC6590611.
- Sturrock, Beattie & Rogers, Jennifer & Sadler, Ross & Ferry, Berne & Chapman, Roger & Lynch, Kate. (2018). Anti-gp210 and anti-sp100 antibody status and ursodeoxycholic acid response in primary biliary cholangitis. Journal of Gastroenterology and Hepatology Research. 7. 2741-2747. 10.17554/j.issn.2224-3992.2018.07.797.
- Aranda, Mario Ariel, Di Carlo María Beatriz . Fosfatasa alcalina: características generales y determinación sérica. Acta Bioquímica Clínica Latinoamericana [en linea]. 2022, 56(3), 257-272[fecha de Consulta 25 de Septiembre de 2023]. ISSN: 0325-2957. Disponible en: https://www.redalyc.org/articulo.oa?id=53573289007
- Chen S, Li MQ, Duan WJ, Li BE, Li SX, Lv TT, et al. Concomitant extrahepatic autoimmune diseases do not compromise the long-term outcomes of primary biliary cholangitis. Hepatobiliary Pancreat Dis Int. 2022;21(6):577–582. doi: 10.1016/j.hbpd.2022.05.009.
- Murillo Perez CF, Ioannou S, Hassanally I, Trivedi PJ, Corpechot C, van der Meer AJ, Lammers WJ, Battezzati PM, Lindor KD, Nevens F, Kowdley KV, Bruns T, Cazzagon N, Floreani A, Mason AL, Gulamhusein A, Ponsioen CY, Carbone M, Lleo A, Mayo MJ, Dalekos GN, Gatselis NK, Thorburn D, Verhelst X, Parés A, Londoño MC, Janssen HLA, Invernizzi P, Vuppalanchi R, Hirschfield GM, Hansen BE, Levy C; Global PBC Study Group. Optimizing therapy in primary biliary cholangitis: Alkaline phosphatase at six months identifies one-year non-responders and predicts survival. Liver Int. 2023 Jul;43(7):1497-1506. doi: 10.1111/liv.15592. Epub 2023 May 8. PMID: 37157905.
Artigo criado em: 17/08/04
Última revisão: 27/09/23



