
Dra. Adriana Maria Alves De Tommaso
INTRODUÇÃO
As glicogenoses são doenças secundárias a um erro no metabolismo, hereditário, o qual resulta de concentrações alteradas de glicogênio no organismo. Existem mais de 10 diferentes tipos, dependendo do defeito enzimático encontrado.
O glicogênio está presente em todas as células animais, sendo mais abundante no fígado e nos músculos. É a forma através da qual armazenamos a glicose da dieta. A glicose chega ao fígado pela veia porta. Quando há necessidade de uso da glicose (situações de stress, jejum), há quebra desse glicogênio por meio de processo enzimático ocorrendo então, liberação da glicose para a circulação sangüínea. Dessa forma, o fígado proporciona a liberação de glicose para vários órgãos, incluindo o cérebro. Quando o glicogênio não consegue ser quebrado devido à deficiência de algumas das enzimas envolvidas, este se acumula no órgão e, a não liberação de glicose para a circulação acarreta uma série de consequências, como veremos a seguir:
GLICOGENOSE TIPO I
É causada pela deficiência da enzima chamada glicose-6-fosfatase. Se subdivide em Ia, Ib e Ic. É o tipo mais comum e o órgão acometido é o fígado. Esta enzima também é encontrada nos músculos, rins e mucosa do intestino delgado. Como esta enzima é a principal responsável pela liberação de glicose para a circulação, é de se esperar que as pessoas com glicogenose tipo I sejam incapazes de manter níveis adequados de glicemia nos períodos de jejum.
Como consequências metabólicas temos:
Hipoglicemia: muitas crianças apresentam, como primeiro sintoma, convulsões secundárias à hipoglicemia. No entanto, a hipoglicemia pode não ser acompanhada de sintomas visto que o cérebro pode fazer uso do ácido láctico como substrato. Estudos têm mostrado que, com o avançar da idade, há uma tendência à diminuição da hipoglicemia. Ainda não se sabe, com certeza, a explicação para esse fato, mas três mecanismos têm sido sugeridos: presença de atividade residual da enzima em alguns indivíduos; presença da enzima amilo-1,6-glicosidase, a qual é responsável pela liberação de cerca de 8-10% da glicose produzida pelo fígado; presença da a-glicosidase ácida que libera todo o glicogênio degradado pelo lisossomo na forma de glicose.
Acidose láctica: o lactato geralmente está aumentado em cerca de quatro vezes o normal. O ácido láctico é produzido nos músculos e hemácias e metabolizado no fígado, sendo desviado para a síntese de ácidos graxos ou gliconeogênese. Seu acúmulo na glicogenose tipo I decorre da sua não utilização na gliconeogênese. Mesmo com o controle da doença, os níveis nunca atingem valores normais.
Aumento do ácido úrico: a hiperuricemia resulta da diminuição da excreção de urato pelo rim (devido competição com ácido láctico) e do aumento na produção de ácido úrico. Como conseqüência podem aparecer cálculos renais, gota e nefropatia.
Hiperlipidemia: algumas características clínicas dessa doença, como obesidade e “face de boneca”, demonstram a deposição anormal de gordura. O proeminente aumento do fígado observado nesses pacientes se deve muito mais à infiltração de gordura do que ao acúmulo de glicogênio. Os níveis séricos de triglicerídios estão bastante elevados, podendo atingir 4000-6000mg/dL. Por outro lado, os níveis de colesterol e fosfolípides estão moderadamente elevados. A hiperlipoproteinemia é principalmente causada pela elevação das frações VLDL e LDL. A alteração do metabolismo dos lipídios está relacionada com a alteração do metabolismo dos carboidratos e conseqüente alteração hormonal. Apesar do perfil lipídico poder sugerir risco para doença coronariana, não há relatos de doença coronariana precoce nesses pacientes.
Desordens hematológicas:
- disfunção das plaquetas (reduzida adesividade e alteração na agregação) levando a um tempo de sangramento prolongado. Como conseqüência, é comum a queixa de sangramentos nasais e tendência para hemorragia durante procedimentos cirúrgicos. O controle da doença corrige essas alterações;
- alteração nos leucócitos polimorfonucleares: neutropenia tem sido descrita no subtipo Ib, podendo atingir valores abaixo de 1500/mm. Também há uma deficiência na função dessas células. Esses fatores contribuem para aumentar a susceptibilidade desses pacientes às infecções.
Alterações gastrointestinais: freqüentemente há o aparecimento de diarréia intermitente, pois como já mencionado, a enzima também ocorre na mucosa do delgado. Tem sido descrita diarréia secundária à sobrecarga de lactose e glicose. Recentemente, há relatos de associação da glicogenose tipo I com doença inflamatória intestinal.
Adenomas e neoplasias hepáticas: os adenomas ocorrem em cerca de 50-70% dos pacientes a partir da segunda e terceira décadas de vida. A patogênese não está muito bem esclarecida. Os dois principais tipos de lesão descritos são o hepatoma e o adenoma hepatocelular com displasia. São detectados por meio da ultra-sonografia ou tomografia computadorizada. Os adenomas podem responder à dieta, diminuindo de tamanho ou tornando-se indetectáveis ou, podem sofrer transformação maligna. Os níveis de a-feto-proteína são úteis na monitorização da progressão do tumor.
- Clínica: Os sinais mais comuns são a hipoglicemia e o aumento da tamanho do fígado. Classicamente, a doença é descrita nos primeiros 28 dias de vida (período neonatal). Os bebês costumam apresentar hipoglicemia após pequenos períodos de jejum ou após infecções. A hipoglicemia se caracteriza por palidez, suor frio, convulsões. Ao exame físico nota-se o aumento hepático, a obesidade troncular, a “face de boneca”. Também há atraso no crescimento estatural. A glicogenose tipo Ib também pode se complicar com infecções piogênicas, gengivoestomatite recorrente e doença inflamatória intestinal.
Diagnóstico: As alterações laboratoriais mais freqüentes são: hipoglicemia após 3-4 horas de jejum (também pode ocorrer com um número menor ou maior de horas), aumento do ácido láctico e do colesterol, aumento de ácido úrico e triglicerídios, aumento de fosfolípides e aumento discreto das enzimas hepáticas (TGO e TGP). A determinação da enzima em amostra de tecido hepático confirma o diagnóstico, mas poucos serviços dispõem desse método. Também pode-se fazer estudos com DNA.
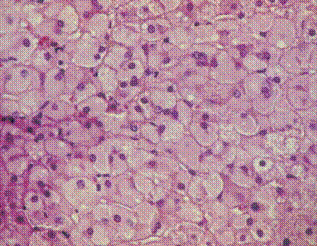
Biópsia hepática evidenciando aspecto típico de célula vegetal do hepatócito.
Tratamento:
- Dieta: amido cru (MaizenaR) a cada 2-4 horas (dependendo do intervalo de tolerância ao jejum) na dose de 1,75-2,5g/kg/dose. Este intervalo também deve ser respeitado durante a noite. O amido deve ser dado, preferencialmente, com água fria. A mistura com água morna ou quente ou limonada, acelera sua hidrólise. O colesterol fica restrito a menos de 200mg/dia. A dieta deve ser isenta de frutose e galactose (leite, frutas, verduras) se não houver melhora dos parâmetros bioquímicos. Geralmente, pequenas doses de frutose e galactose são bem toleradas. Nos casos mais graves, há necessidade de utilização de sonda nasogástrica.
- Transplante Hepático: pode ser considerado nos casos que não respondem ao manejo dietético e necessitam freqüentes internações.
- Hiperuricemia: o alopurinol pode ser usados nos casos em que o manejo dietético não é suficiente para controlar os níveis de ácido úrico.
- Disfunção plaquetária: a infusão de DDAVP pode ser útil para os casos de hemorragia ou antes de procedimentos cirúrgicos.
GLICOGENOSE TIPO III
- Também chamada doença de Cori, é causada pela deficiência da enzima amilo-1,6-glicosidase a qual é expressa em muitos tecidos. Clinicamente, é muito parecida com a glicogenose tipo I. Apresenta hipoglicemia, aumento do fígado e retardo do crescimento. Durante a Segunda década de vida, a velocidade de crescimento aumenta, o fígado diminui e a hipoglicemia melhora tanto que, muitos adultos toleram várias horas de jejum.
- Usualmente, não há elevação dos níveis de triglicérides, mas freqüentemente, o colesterol está elevado.
- Há, também, comprometimento da musculatura e evidências de cardiomiopatia têm sido encontradas.
- O tratamento é semelhante ao da glicogenose tipo I, sendo que esses pacientes se beneficiam de alta ingesta proteica.
GLICOGENOSE TIPO IV
- Também chamada doença de Andersen, é causada pela deficiência da enzima amilo-1,4-1,6-glicosidase.
- Acomete, principalmente, o fígado. O quadro clínico é de hepatoesplenomegalia (aumento do fígado e do baço) e alterações neuromusculares variadas.
- Não há tratamento eficaz. O transplante de fígado é a única modalidade curativa.
GLICOGENOSE TIPO V
- Também chamada doença de MacArdle, é causada pela deficiência da enzima fosforilase muscular.
- Acomete os músculos. O quadro clínico é de dor muscular (cãimbras) induzida pelo exercício e fraqueza progressiva, algumas vezes associada a mioglobinúria.
GLICOGENOSE TIPO VI
- Também chamada doença de Hers, é causada pela deficiência da enzima fosforilase hepática.
- Clínica e laboratório: hepatomegalia, leve retardo de crescimento, boa tolerância ao jejum, elevação de triglicérides, colesterol e ácido úrico.
- Tratamento: dieta semelhante ao tipo III, pouca necessidade de tratamento.
GLICOGENOSE TIPO VII
- Causada pela deficiência da enzima fosfofrutoquinase.
- A clínica é semelhante ao tipo V, acrescentando-se hemólise.
GLICOGENOSE TIPO IX
- Causada pela deficiência da enzima fosforilase quinase.
- Dividida em subtipos A, B e C.
- Acomete o fígado e, talvez, o músculo. A clínica é semelhante ao tipo VI. O subtipo C também acomete as células sanguíneas.
GLICOGENOSE TIPO X
- Causada pela deficiência do AMP cíclico dependente de quinase.
- Acomete fígado e músculos. A clínica se caracteriza por hepatomegalia.
Artigo criado em: 2003
Última revisão: 2003


